Page Last Updated: April 8, 2026
Genomics Workflows🔗
IDAT to raw PLINK files🔗
- Raw IDAT generated by vendor and placed within HST secure computing environment
- Workflow run which takes raw IDAT files:
- Normalization
- Genotyping
- Final report
- PLINK conversion
PLINK Files🔗
PLINK files: .bed, .bim, .fam - Space delimited with six columns:🔗
- Family ID (FID)
- Within-family ID (IID) - i.e. FID plus relationship in family C = child, M = mother
- Within-family ID of father
- Within-family ID of mother
- Sex
- Phenotype
batch.info - Tab delimited with 3 columns:🔗
- IID (character, 10 digits + 1 letter either M or C)
- plate_number (float)
- visit (character)
.fam and batch.info have the same ordering of IID column🔗
Genomic Derivatives🔗
- Principal components -
.eigenvec,.eigenval - Inferred Sex
- Inferred Ancestry
Prerelease staging file structure🔗
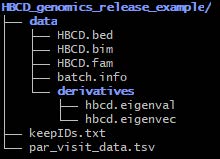
- Example directory structure for internal data storage
- This data is not ready for public as it is not deidentified!!!
- Only files in the “data” subdirectory are to be shared other files are supporting files in generating the release data
- For each release files will be placed here before running deidentification
/projects/standard/basu_hbcd/shared/HBCD_genomics_release_<release details>- Newly associated files that are not included release (such as par_visit_data.tsv) will be in the firs level of this directory
- Data to be released will be in the “data” directory
- QC’d genomic data will be stored as Plink1 binary (
.bed/.bim/.fam) format in the first level of the “data” directory - Derivatives associated with genomics release will be stored in “data/derivatives”
Generation of release ready PLINK files🔗
- Sample-Level QC (match with .fam file generated by Sampled) - call rate, sex check, family relatedness, PCA
- Sample-Level QC (match with the LORIS demographic file) - sex check, relatedness, ancestry
- Variant level QC - Batch effects
- To ensure that PLINKs are ready for a given release they must be filtered with the
par_visit_data.tsvused for the release
Deidentification, folder structure, and push to release bucket🔗
- Use a recent mapping file present within
s3://midb-hbcd-main-pr-deidentification-list- maps pscid to release candid - Script within https://github.umn.edu/MIDB-IG/data-release/tree/genomics-de-id named
deidentify_genomics.py- Will de-identify
.fam, and batch info file based on provided mapping file - Copy de-identified
.famand batch info file along with.bedand.bimfiles into a separate folder - Folder designed to emulate ABCD directory structure (i.e.
<output folder>/concat/genetics/genotype_microarray/GDA)
- Will de-identify
- Once produced a validation script will be run to ensure that appropriate participants are present and that all are de-id’ed
- Confirming that FID columns are completely numeric
- Confirming that IID columns are numeric plus a single character (1 of either “M” or “C”)
- Confirming that the FID and IID columns are of fixed width (9 and 10 characters respectively
- Once validated, de-id’ed release data will be pushed to pre-release bucket - i.e.BR:
s3://midb-hbcd-lasso-hdcc-qc-br/br<number>